Publications
Group highlights
At the end of this page, you can find the full list of publications.
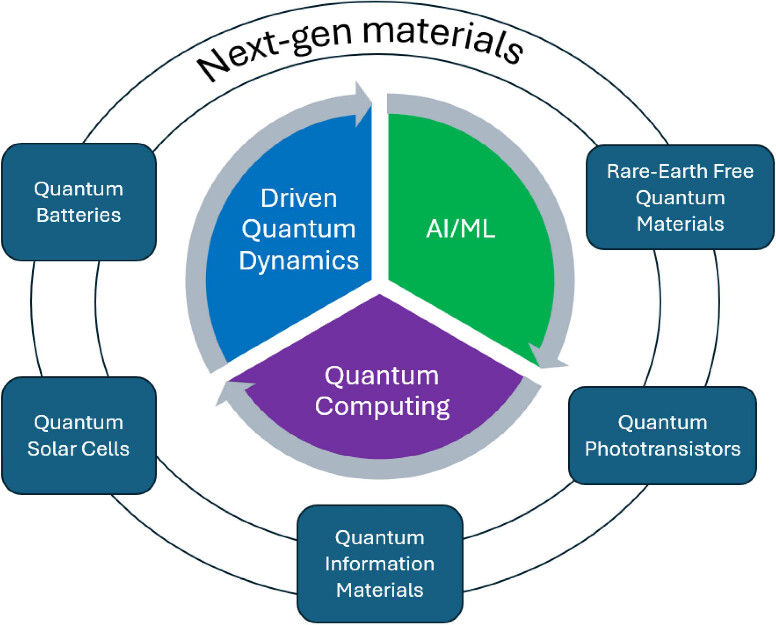
Recent advances in next-generation materials design leverage synergistic approaches combining AI-driven synthesis, robotics, and quantum computing to accelerate the discovery of quantum materials with enhanced properties and novel functionalities for applications such as quantum batteries, solar cells, and phototransistors.
O. S. Akanbi, J. P. Shannon, J. Delhommelle, C. Desgranges
J. Phys. Chem. Lett. 16, 45 (2025)
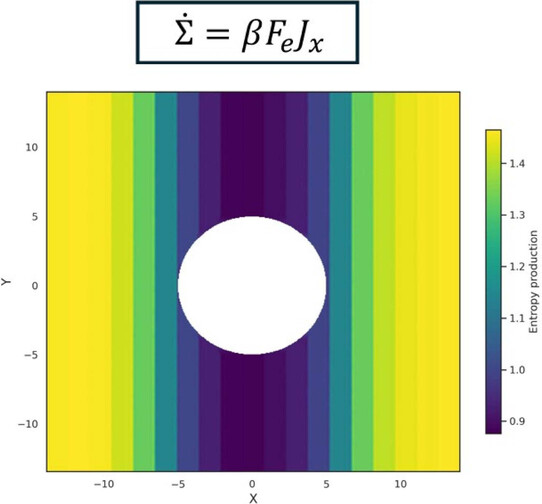
The work introduces a framework that decomposes global entropy production into local contributions by analyzing heat flows and fluxes, enabling spatial maps of dissipation in driven and active fluids and revealing that local entropy production obeys a local fluctuation theorem with correlations of opposite sign for stochastic and deterministic contributions in active matter.
C. Desgranges, J. Delhommelle
J. Phys. Chem. Lett. 16, 11405 (2025)
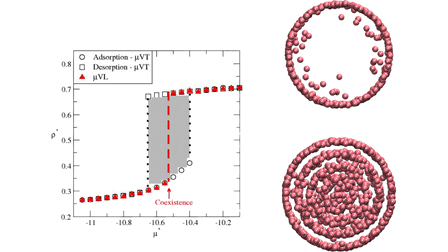
Here, we leverage simulations in an adiabatic statistical ensemble, known as adiabatic grand-isochoric ensemble (μ, V, L) ensemble, to reach equilibrium states with a greater efficiency than its isothermal counterpart, i.e., simulations in the grand-canonical ensemble.
C. Desgranges, J. Delhommelle
J. Phys. Chem. 161, 104104 (2024) (Editors’ Pick)

This book employs an analogy between social groups and molecular assemblies to introduce statistical mechanics, machine learning, and data science concepts, demonstrating how molecular features, interactions, and “communication” processes, akin to polling and social networking, enable prediction of properties and collective behavior in systems for biological, environmental, and energy applications.
C. Desgranges, J. Delhommelle
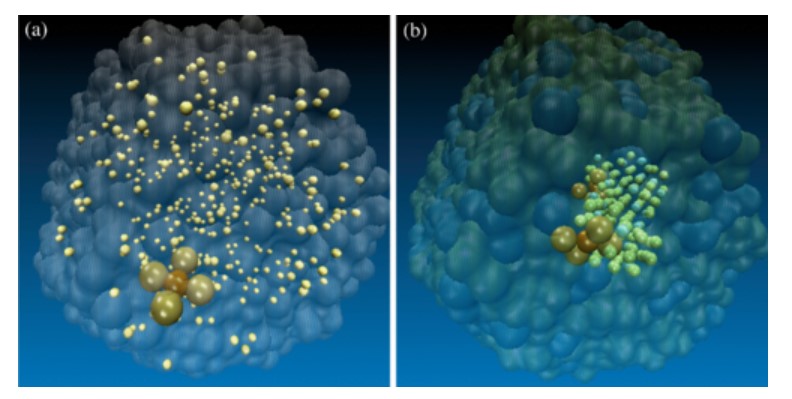
Crystallization often proceeds through successive stages that lead to a gradual increase in organization. Using molecular simulation, we determine the nucleation pathway for solid solutions of copper and gold. We identify a new nucleation mechanism (liquid→𝐿12 precursor→solid solution) involving a chemically ordered intermediate that is more organized than the end product.
C. Desgranges, J. Delhommelle
Phys. Rev. Lett. 123, 195701 (2019)

We combine ML with Monte Carlo simulations to study the crystal nucleation process. By taking entropy as a reaction coordinate and using the umbrella sampling technique, we are able to determine the Gibbs free energy profile for the crystal nucleation process. The approach developed here can be readily extended to molecular systems and complex fluids, and is especially promising for the study of entropy-driven processes.
C. Desgranges, J. Delhommelle
Phys. Rev. E 98, 063307 (2018)
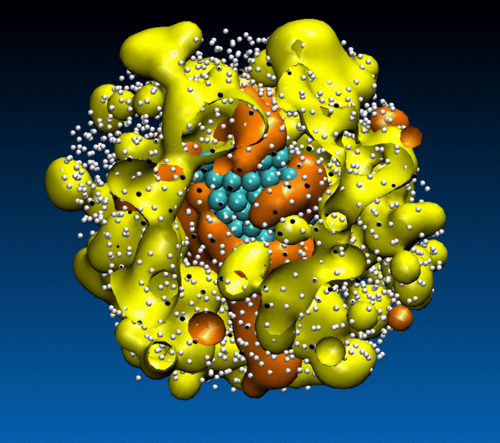
Using molecular simulations, we shed light on the mechanism underlying crystal nucleation in metal alloys and unravel the interplay between crystal nucleation and glass transition, as the conditions of crystallization lie close to this transition.
C. Desgranges, J. Delhommelle
Phys. Rev. Lett. 120, 115701 (2018)
See Cover of PRL

Using molecular simulation, we shed light on the coupling between the two nonequilibrium processes of demixing and crystallization in mixtures of fully miscible, size-matched, liquid metals.
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 136, 23, 8145–8148 (2014)
See Cover of JACS
See Revealing the Magic behind Nanoscale Crystallization by Jenny Morber
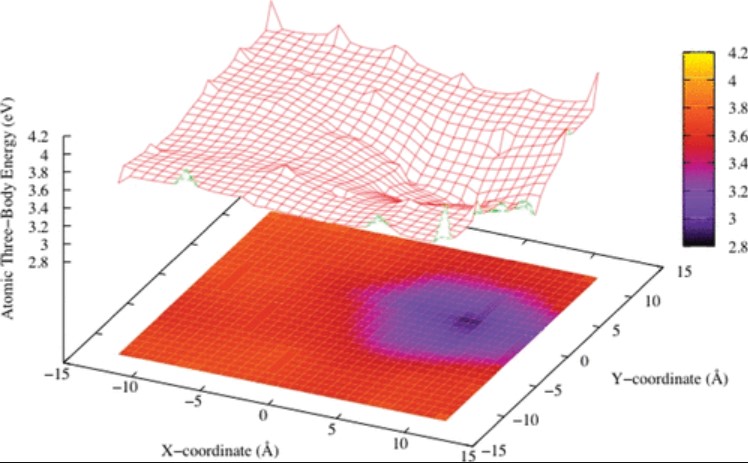
When undercooled at a temperature 20% below the melting point, a silicon melt is under the form of the highly coordinated, high-density liquid (HDL) polymorph. We find that crystallization starts with the formation, within the HDL liquid, of a nanosized droplet of the least stable liquid polymorph, known as the almost tetracoordinated low-density liquid (LDL) polymorph.
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 133, 9, 2872–2874 (2011)

We use molecular dynamics simulations to shed light on polymorph selection during the crystallization of the Lennard-Jones fluid. By varying pressure at fixed supercooling, we form large crystallites either of the stable face centered cubic form or of the metastable body centered cubic form and even fine-tune the fractions of stable and metastable polymorphs in the crystallite.
C. Desgranges, J. Delhommelle
Phys. Rev. Lett. 98, 235502 (2007)
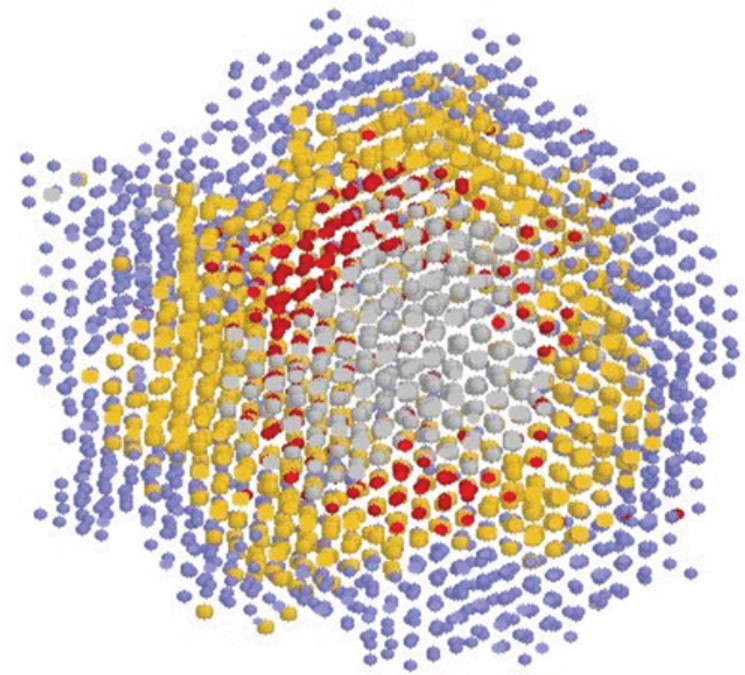
Body-centered cubic clusters, which usually form during the nucleation of simple fluids, are not observed during the crystallization of Al. Throughout nucleation and growth, Al nuclei are always strongly faceted, in sharp contrast with the spherical crystallites observed for simple fluids.
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 129, 22, 7012–7013 (2007)
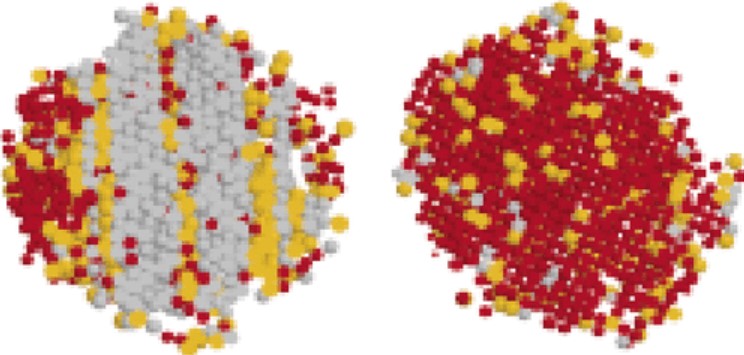
We use molecular simulations to study polymorph selection during the crystallization of charge-stabilized colloidal suspension. By modifying the conditions of crystallization, we invert the stability of two polymorphs and induce the formation of crystallites whose structure is predominantly that of the stable polymorph.
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 128, 47, 15104–15105 (2006)
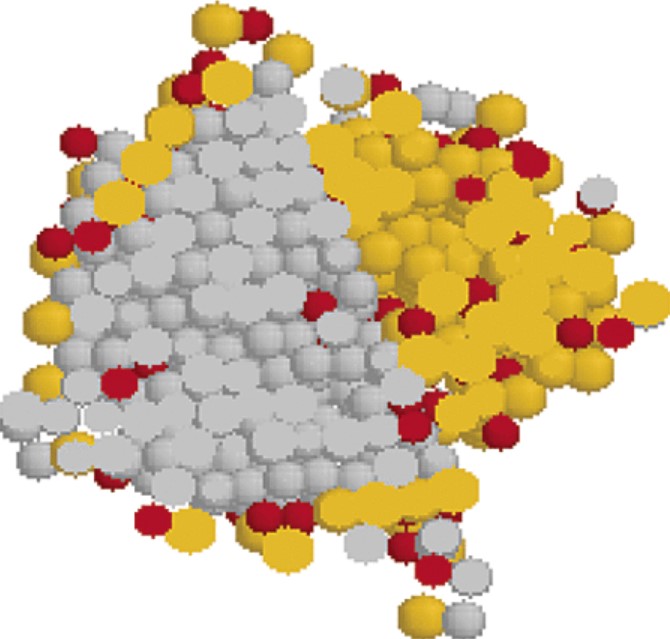
We use molecular simulations to study the early stages of crystallization in a supercooled liquid of Lennard-Jones particles. We observe the onset of concomitant polymorphism and demonstrate that this phenomenon results from the cross-nucleation of a metastable polymorph on the stable polymorph. We also show that cross-nucleation is selective as it only takes place between polymorphs of almost equivalent free energy.
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 128, 32, 10368–10369 (2006)
Full List of publications
Mini Review: Synergizing Driven Quantum Dynamics, AI, and Quantum Computing for Next-Gen Materials Science
O. S. Akanbi, J. P. Shannon, J. Delhommelle, C. Desgranges
J. Phys. Chem. Lett. 16, 45 (2025)
Mapping Local Dissipation and Entropy Production in Complex and Active Fluids
C. Desgranges, J. Delhommelle
J. Phys. Chem. Lett. 16, 11405 (2025)
Deciphering the complexities of crystalline state (s) with molecular simulations
C. Desgranges, J. Delhommelle,
Commun. Chem. 8, 281 (2025)
Advancing the design of gold nanomaterials with machine-learned potentials
K. Sajini, C. Desgranges, J. Delhommelle,
Nano Express 6, 022001 (2025)
Accelerated convergence via adiabatic sampling for adsorption and desorption processes
C. Desgranges, J. Delhommelle
J. Phys. Chem. 161, 104104 (2024) (Editors’ Pick)
Molecular Networking: Statistical Mechanics in the Age of AI and Machine Learning
C. Desgranges, J. Delhommelle
CRC Press, 2024
Microswimmers under the spotlight: interplay between agents with different levels of activity
C. Desgranges, M. Ferrari, P. M. Chaikin, S. Sacanna, M. E. Tuckerman, J. Delhommelle
Soft Matter 19, 7334 (2023)
Designing, synthesizing, and modeling active fluids
I. Essafri, B. Ghosh, C. Desgranges, J. Delhommelle
Phys. Fluids 34, 071301 (2022)
Entropy determination for mixtures in the adiabatic grand-isobaric ensemble
C. Desgranges, J. Delhommelle
J. Chem. Phys. 156, 084113 (2022)
Machine-Learned Free Energy Surfaces for Capillary Condensation and Evaporation in Mesopores
C. Desgranges, J. Delhommelle
Entropy 24, 97 (2022)
Entropy scaling close to criticality: From simple to metallic systems
C. Desgranges, J. Delhommelle
Phys. Rev. E 103, 052102 (2021)
Towards a machine learned thermodynamics: exploration of free energy landscapes in molecular fluids, biological systems and for gas storage and separation in metal–organic frameworks
C. Desgranges, J. Delhommelle
Molecular Systems Design & Engineering 6, 52 (2021)
Entropy production in model colloidal suspensions under shear via the fluctuation theorem
C. Desgranges, J. Delhommelle
J. Phys. Chem. 153, 224113 (2020)
Entropy in Molecular Fluids: Interplay between Interaction Complexity and Criticality
C. Desgranges, J. Delhommelle
J. Phys. Chem. B 124, 11463 (2020)
Can Ordered Precursors Promote the Nucleation of Solid Solutions?
C. Desgranges, J. Delhommelle
Phys. Rev. Lett. 123, 195701 (2019)
Crystal nucleation along an entropic pathway: Teaching liquids how to transition
C. Desgranges, J. Delhommelle
Phys. Rev. E 98, 063307 (2018)
Unusual Crystallization Behavior Close to the Glass Transition
C. Desgranges, J. Delhommelle
Phys. Rev. Lett. 120, 115701 (2018)
Free energy calculations along entropic pathways. II. Droplet nucleation in binary mixtures
C. Desgranges, J. Delhommelle
J. Chem. Phys. 145, 234505 (2016)
Effect of the Composition on the Free Energy of Crystal Nucleation for CuPd Nanoalloys
C. Desgranges, J. Delhommelle
J. Phys. Chem. C 120, 48, 27657–27664 (2016)
Free energy calculations along entropic pathways. I. Homogeneous vapor-liquid nucleation for atomic and molecular systems
C. Desgranges, J. Delhommelle
J. Chem. Phys. 145, 204112 (2016)
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. V. Impact of an electric field on the thermodynamic properties and ideality contours of water
C. Desgranges, J. Delhommelle
J. Chem. Phys. 145, 184504 (2016)
Ideality contours and thermodynamic regularities in supercritical molecular fluids
C. Desgranges, A. Margo, J. Delhommelle
Chemical Physics Letters 658, 37-42 (2016)
Impact of Friedel oscillations on vapor-liquid equilibria and supercritical properties in two and three dimensions
C. Desgranges, L. Huber, J. Delhommelle
Phys. Rev. E 94, 012612 (2016)
Scaling Laws and Critical Properties for fcc and hcp Metals
C. Desgranges, L. Huber, J. Delhommelle
J. Phys. Chem. B 120, 23, 5255–5261 (2016)
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. IV. Performance of many-body force fields and tight-binding schemes for the fluid phases of silicon
C. Desgranges, J. Delhommelle
J. Chem. Phys. 144, 124510 (2016)
Many-Body Effects on the Thermodynamics of Fluids, Mixtures, and Nanoconfined Fluids
C. Desgranges, J. Delhommelle
J. Chem. Theory Comput. 11, 11, 5401–5414 (2015)
A new force field for H2S and its binary and ternary mixtures with CO2 and CH4
A.N. Owen, C. Desgranges, J. Delhommelle
Fluid Phase Equilibria, 402, 69-77 (2015)
Adsorption and diffusion of the antiparkinsonian drug amantadine in carbon nanotubes
E. Hicks, C. Desgranges, J. Delhommelle
Molecular Simulation, 40(7–9), 656–663 (2014)
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. III. Impact of combining rules on mixtures properties
C. Desgranges, J. Delhommelle
J. Chem. Phys. 140, 104109 (2014)
Thermodynamics of Phase Coexistence and Metal–Nonmetal Transition in Mercury: Assessment of Effective Potentials via Expanded Wang–Landau Simulations
C. Desgranges, J. Delhommelle
J. Phys. Chem. B 118, 11, 3175–3182 (2014)
Unraveling the Coupling between Demixing and Crystallization in Mixtures
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 136, 23, 8145–8148 (2014)
Adsorption of hydrogen in covalent organic frameworks using expanded Wang–Landau simulations
A.R.V. Koenig, C. Desgranges, J. Delhommelle
Molecular Simulation, 40(1–3), 71–79. (2013)
Characterization and Comparison of the Performance of IRMOF-1, IRMOF-8, and IRMOF-10 for CO2 Adsorption in the Subcritical and Supercritical Regimes
J. M. Hicks, C. Desgranges, J. Delhommelle
J. Phys. Chem. C 116, 43, 22938–22946 (2012)
Numerical estimate for boiling points via Wang–Landau simulations
T. Aleksandrov, C. Desgranges, J. Delhommelle
Molecular Simulation, 38(14–15), 1265–1270 (2012)
Wang–Landau configurational bias Monte Carlo simulations: vapour–liquid equilibria of alkenes
K. Ndumbe Ngale, C. Desgranges, J. Delhommelle
Molecular Simulation, 38(8–9), 653–658 (2012)
Prediction of critical properties for naphthacene, triphenylene and chrysene by Wang–Landau simulations
C. Desgranges, K. Ndumbe Ngale, J. Delhommelle
Fluid Phase Equilibria, 322–323, 92-96, (2012)
Prediction of critical properties for naphthacene, triphenylene and chrysene by Wang–Landau simulations
C. Desgranges, K. Ndumbe Ngale, J. Delhommelle
Fluid Phase Equilibria, 322–323, 92-96, (2012)
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. II. Adsorption of atomic and molecular fluids in a porous material
C. Desgranges, J. Delhommelle
J. Chem. Phys. 136, 184108 (2012)
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. I. Thermodynamic properties in the bulk and at the liquid-vapor phase boundary
C. Desgranges, J. Delhommelle
J. Chem. Phys. 136, 184107 (2012)
Polymorph selection during the crystallization of iron under the conditions of Earth’s inner core
J. Persson, C. Desgranges, J. Delhommelle
Chem. Phys. Lett. 57-61 (2011)
Role of Liquid Polymorphism during the Crystallization of Silicon
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 133, 9, 2872–2874 (2011)
Crystal nucleation and growth in Pd–Ni alloys: a molecular simulation study
K. Watson, S.E. Tatsinkou Nguelo, C. Desgranges, J. Delhommelle
CrystEngComm, 13, 1132-1140 (2011)
Optimisation of multiple time-step hybrid Monte Carlo Wang–Landau simulations in the isobaric–isothermal ensemble for the determination of phase equilibria
C. Desgranges, E.A. Kastl, T. Aleksandrov, J. Delhommelle
Molecular Simulation, 36(7–8), 544–551 (2010)
Phase equilibria of polyaromatic hydrocarbons by hybrid Monte Carlo Wang–Landau simulations
C. Desgranges, J.M. Hicks, A. Magness, J. Delhommelle
Molecular Physics, 108, 151–158 (2010)
Vapor–liquid equilibria of copper using hybrid Monte Carlo Wang—Landau simulations
T. Aleksandrov, C. Desgranges, J. Delhommelle
Fluid Phase Equilibria, 287, 79-83 (2010)
Nucleation and growth of nanoparticles from the supersaturated vapor and from the undercooled liquid: A molecular simulation study
K. N. Ngale, C. Desgranges, J. Delhommelle
J. Chem. Phys. 131, 244515 (2009)
Phase equilibria of molecular fluids via hybrid Monte Carlo Wang–Landau simulations: Applications to benzene and n-alkanes
C. Desgranges, J. Delhommelle
J. Chem. Phys. 130, 244109 (2009)
Universal scaling law for energy and pressure in a shearing fluid
C. Desgranges, J. Delhommelle
Phys. Rev. E 79, 052201 (2009)
Accurate determination of normal stress differences via transient-time correlation function – non-equilibrium molecular dynamics (TTCF–NEMD) simulations
C. Desgranges, J. Delhommelle
Molecular Simulation, 35(5), 405–408 (2009)
Molecular Simulation of the Nucleation and Growth of Gold Nanoparticles
C. Desgranges, J. Delhommelle
J. Phys. Chem. C 113, 9, 3607–3611 (2009)
Rheology of liquid fcc metals: Equilibrium and transient-time correlation-function nonequilibrium molecular dynamics simulations
C. Desgranges, J. Delhommelle
Phys. Rev. B 78, 184202 (2008)
Shear viscosity of liquid copper at experimentally accessible shear rates: Application of the transient-time correlation function formalism
C. Desgranges, J. Delhommelle
J. Chem. Phys. 128, 084506 (2008)
Molecular simulation of transport in nanopores: Application of the transient-time correlation function formalism
C. Desgranges, J. Delhommelle
Phys. Rev. E 77, 027701 (2008)
Crystallization mechanisms for supercooled liquid Xe at high pressure and temperature: Hybrid Monte Carlo molecular simulations
C. Desgranges, J. Delhommelle
Phys. Rev. B 77, 054201 (2008)
Estimating the conductivity of a nanoconfined liquid subjected to an experimentally accessible external field
C. Desgranges, J. Delhommelle
Molecular Simulation, 34(2), 177–181 (2008)
Viscosity of liquid iron under high pressure and high temperature: Equilibrium and nonequilibrium molecular dynamics simulation studies
C. Desgranges, J. Delhommelle
Phys. Rev. B 76, 172102 (2007)
Polymorph Selection during the Crystallization of Softly Repulsive Spheres: The Inverse Power Law Potential
C. Desgranges, J. Delhommelle
J. Phys. Chem. B 111, 42, 12257–12262 (2007)
Molecular simulation of the crystallization of aluminum from the supercooled liquid
C. Desgranges, J. Delhommelle
J. Chem. Phys. 127, 144509 (2007)
Controlling Polymorphism during the Crystallization of an Atomic Fluid
C. Desgranges, J. Delhommelle
Phys. Rev. Lett. 98, 235502 (2007)
Molecular Insight into the Pathway to Crystallization of Aluminum
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 129, 22, 7012–7013 (2007)
Molecular Simulation of Cross-Nucleation between Polymorphs
C. Desgranges, J. Delhommelle
J. Phys. Chem. B 111, 6, 1465–1469 (2007)
Polymorph selection during the crystallization of Yukawa systems
C. Desgranges, J. Delhommelle
J. Chem. Phys. 126, 054501 (2007)
Insights into the Molecular Mechanism Underlying Polymorph Selection
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 128, 47, 15104–15105 (2006)
Molecular Mechanism for the Cross-Nucleation between Polymorphs
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc. 128, 32, 10368–10369 (2006)